
2. Typical usage for DNAseq®¶
One of the typical uses of Sentieon® Genomics software is to perform the bioinformatics pipeline for DNA analysis recommended in the Broad institute best practices described in https://www.broadinstitute.org/gatk/guide/best-practices. Fig. 2.1 illustrates such a typical bioinformatics pipeline.
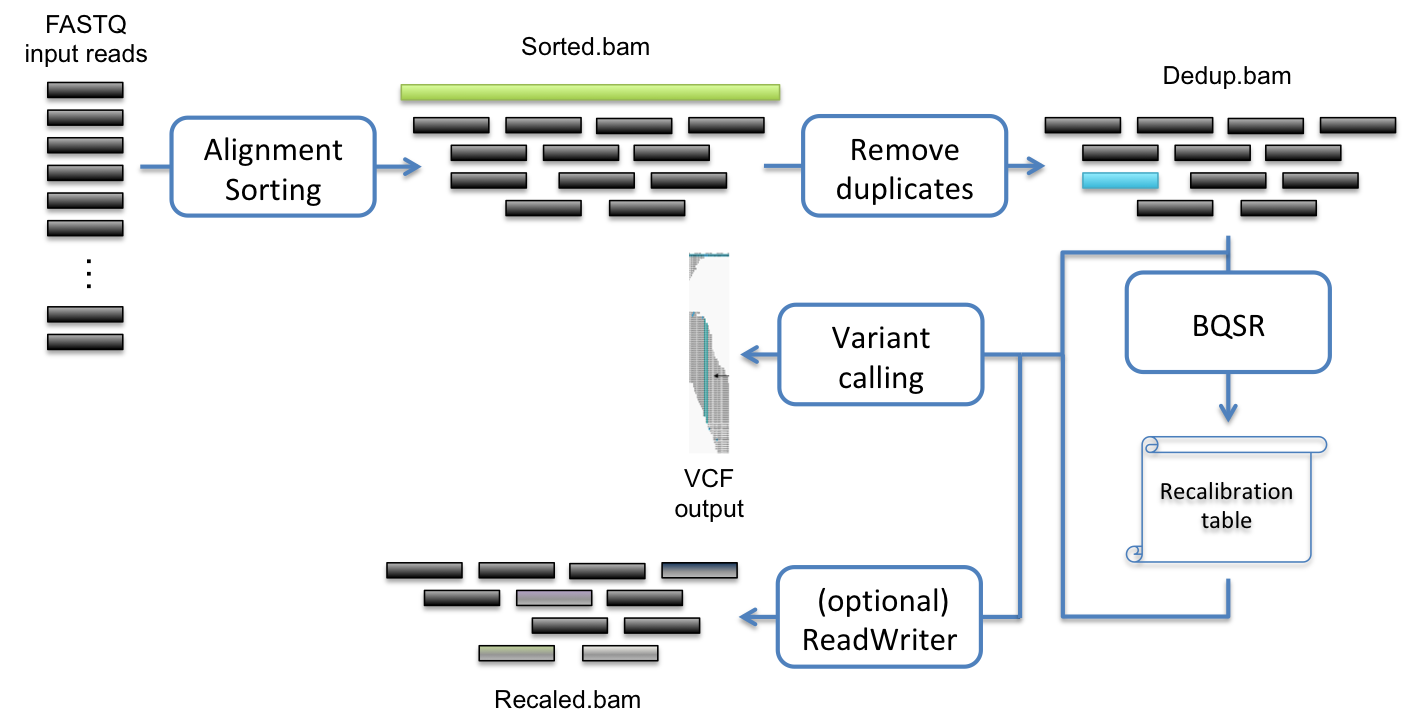
Fig. 2.1 Recommended bioinformatics pipeline for DNA variant calling analysis
2.1. General¶
In this bioinformatics pipeline you will need the following inputs:
The FASTA file containing the nucleotide sequence of the reference genome corresponding to the sample you will analyze. The reference data needs to be pre-processed such that the data specified in Table 2.1 is available to the software. You can refer to Preparing reference file for use for instructions on how to generate the required files.
Table 2.1 Data requirements for the reference nucleotide sequence¶ Data Description .dict Dictionary file .fasta Reference sequence file .fasta.amb BWA index file .fasta.ann BWA index file .fasta.bwt BWA index file .fasta.fai Index file .fasta.pac BWA index file .fasta.sa BWA index file One or multiple FASTQ files containing the nucleotide sequence of the sample to be analyzed. These files contain the raw reads from the DNA sequencing. The software supports inputting FASTQ files compressed using GZIP. The software only supports files containing quality scores in Sanger format (Phred+33).
(Optional) The Single Nucleotide Polymorphism database (dbSNP) data that you want to include in the pipeline. The data is used in the form of a VCF file; you can use a VCF file compressed with bgzip and indexed.
(Optional) Multiple collections of known sites that you want to include in the pipeline. The data is used in the form of a VCF file; you can use a VCF file compressed with bgzip and indexed.
The following steps compose the typical bioinformatics pipeline:
- Map reads to reference: This step aligns the reads contained in the FASTQ files to map to a reference genome contained in the FASTA file. This step ensures that the data can be placed in context.
- Calculate data metrics: This step produces a statistical summary of the data quality and the pipeline data analysis quality.
- Remove or mark duplicates: This step detects reads indicative that the same DNA molecules were sequenced several times. These duplicates are not informative and should not be counted as additional evidence.
- (Optional) Base quality score recalibration (BQSR): This step modifies the quality scores assigned to individual read bases of the sequence read data. This action removes experimental biases caused by the sequencing methodology.
- Variant calling: This step identifies the sites where your data displays variation relative to the reference genome, and calculates genotypes for each sample at that site.
2.2. Step by step usage for DNAseq®¶
2.2.1. Map reads to reference¶
A single command is run to efficiently perform the alignment using BWA, and the creation of the BAM file and the sorting using Sentieon® software:
(sentieon bwa mem -R '@RG\tID:GROUP_NAME\tSM:SAMPLE_NAME\tPL:PLATFORM' \
-t NUMBER_THREADS REFERENCE SAMPLE [SAMPLE2] || echo -n 'error' ) \
| sentieon util sort -r REFERENCE -o SORTED_BAM -t NUMBER_THREADS --sam2bam -i -
Inputs and options for BWA are described in its manual.
Alternatively, you can use other aligners that produce a file following the SAM format in stdout and replace the BWA portion of the command.
The following inputs are required for the command:
- GROUP_NAME: Readgroup identifier that will be added to the
readgroup header line. The
RG:IDneeds to be unique among all the datasets that you plan on using, which is important when working with multiple input files as described in Section 8.2, or when performing a Tumor-Normal analysis as described in Section 5. - SAMPLE_NAME: name of the sample that will be added to the readgroup header line.
- PLATFORM: name of the sequencing platform used to sequence the DNA. Possible options are ILLUMINA when the fastq files have been produced in an IlluminaTM machine; IONTORRENT when the fastq files have been produced in a Life TechnologiesTM Ion-TorrentTM machine; ELEMENT when the fastq files have been produced in an Element BiosciencesTM machine; DNBSEQ when the fastq files have been produced in a MGITM machine; ULTIMA when the fastq files have been produced in an Ultima GenomicsTM machine.
- NUMBER_THREADS: the number of computer threads that will be used in the calculation. We recommend that the number does not exceed the number of computing cores available in your system. We recommend that you use the same number of threads for both BWA and for the util binary.
- REFERENCE: the location of the reference FASTA file. You should make sure that all additional reference data specified in Table 2.1 is available in the same location with consistent naming.
- SAMPLE: the location of the sample FASTQ file. If the data comes from pair ended sequencing technology, you will also need to input SAMPLE2 as the corresponding pair sample FASTQ file.
- SORTED_BAM: the location and filename of the sorted mapped BAM output file. A corresponding index file (.bai) will be created.
BWA will produce slightly different results depending on the number of
threads used in the command. This is due to the fact that BWA computes
the insert size distribution on a chunk, whose size is dependent on the
number of threads. To guarantee that the results are independent of the
number of threads used, you should fix the chunk size in bases using
option -K 10000000.
2.2.2. Calculate data metrics¶
A single command is run to generate 5 statistical summaries of the data quality and the pipeline data analysis quality results:
sentieon driver -t NUMBER_THREADS -r REFERENCE -i SORTED_BAM \
--algo GCBias --summary GC_SUMMARY_TXT GC_METRIC_TXT \
--algo MeanQualityByCycle MQ_METRIC_TXT \
--algo QualDistribution QD_METRIC_TXT \
--algo InsertSizeMetricAlgo IS_METRIC_TXT \
--algo AlignmentStat ALN_METRIC_TXT
Four commands are run to generate the plots from the statistical summaries:
sentieon plot GCBias -o GC_METRIC_PDF GC_METRIC_TXT
sentieon plot MeanQualityByCycle -o MQ_METRIC_PDF MQ_METRIC_TXT
sentieon plot QualDistribution -o QD_METRIC_PDF QD_METRIC_TXT
sentieon plot InsertSizeMetricAlgo -o IS_METRIC_PDF IS_METRIC_TXT
The following inputs are required for the commands:
- NUMBER_THREADS: the number of computer threads that will be used in the calculation. We recommend that the number does not exceed the number of computing cores available in your system.
- REFERENCE: the location of the reference FASTA file. You should make sure that the reference is the same as the one used in the mapping stage.
- SORTED_BAM: the location where the previous mapping stage stored the result.
- GC_SUMMARY_TXT: the location and filename of the GC bias metrics summary results output file.
- GC_METRIC_TXT: the location and filename of the GC bias metrics results output file.
- MQ_METRIC_TXT: the location and filename of the mapping quality metrics results output file.
- QD_METRIC_TXT: the location and filename of the quality/depth metrics results output file.
- IS_METRIC_TXT: the location and filename of the insertion size metrics results output file.
- ALN_METRIC_TXT: the location and filename of the alignment metrics results output file.
- GC_METRIC_PDF: the location and filename of the GC bias metrics report output file.
- MQ_METRIC_PDF: the location and filename of the mapping quality metrics report output file.
- QD_METRIC_PDF: the location and filename of the quality/depth metrics report output file.
- IS_METRIC_PDF: the location and filename of the insertion size metrics report output file.
2.2.3. Remove or mark duplicates¶
Two individual commands are run to remove or mark duplicates on the BAM file
after alignment and sorting. The first command collects read
information, and the second command performs the deduplication; the option
--rmdup controls whether the duplicated reads are removed, if the option is
present, or only marked as duplicated.
sentieon driver -t NUMBER_THREADS -i SORTED_BAM \
--algo LocusCollector --fun score_info SCORE.gz
sentieon driver -t NUMBER_THREADS -i SORTED_BAM \
--algo Dedup [--rmdup] --score_info SCORE.gz \
--metrics DEDUP_METRIC_TXT DEDUPED_BAM
The following inputs are required for the commands:
- NUMBER_THREADS: the number of computer threads that will be used in the calculation. We recommend that the number does not exceed the number of computing cores available in your system.
- SORTED_BAM: the location where the previous mapping stage stored the result.
- SCORE.gz: the location and filename of the temporary score output file. Make sure that the same file is used for both commands.
- DEDUP_METRICS_TXT: the location and filename of the dedup metrics results output file.
- DEDUPED_BAM: the location and filename of the deduped BAM output file. A corresponding index file (.bai) will be created.
2.2.4. Base quality score recalibration (BQSR; optional)¶
A single command is run to calculate the required modification of the quality scores assigned to individual read bases of the sequence read data; the actual recalibration is applied during the variant calling stage.
sentieon driver -t NUMBER_THREADS -r REFERENCE \
-i DEDUPED_BAM --algo QualCal [-k KNOWN_SITES] RECAL_DATA.TABLE
Three commands are run to apply the recalibration and create a report on the base quality sore recalibration. The first command applies the recalibration to calculate the post calibration data table and additionally apply the recalibration on the BAM file, the second one creates the data for plotting; the third command plots the calibration data tables, both pre and post, into graphs in a pdf.
sentieon driver -t NUMBER_THREADS -r REFERENCE -i DEDUPED_BAM \
-q RECAL_DATA.TABLE --algo QualCal [-k KNOWN_SITES] \
RECAL_DATA.TABLE.POST [--algo ReadWriter RECALIBRATED_BAM]
sentieon driver -t NUMBER_THREADS --algo QualCal --plot \
--before RECAL_DATA.TABLE --after RECAL_DATA.TABLE.POST RECAL_RESULT.CSV
sentieon plot QualCal -o BQSR_PDF RECAL_RESULT.CSV
The following inputs are required for the command:
- NUMBER_THREADS: the number of computer threads that will be used in the calculation. We recommend that the number does not exceed the number of computing cores available in your system.
- REFERENCE: the location of the reference FASTA file. You should make sure that the reference is the same as the one used in the mapping stage.
- DEDUPED_BAM: the location where the previous deduping stage stored the result.
- RECAL_DATA.TABLE: the location and filename of the recalibration table.
- RECAL_DATA.TABLE.POST: the location and filename of the temporary post recalibration table
- RECAL_RESULT.CSV: the location and filename of the temporary recalibration results output file used for plotting.
- BQSR_PDF: the location and filename of the BSQR results output file.
The following inputs are optional for the command:
- KNOWN_SITES: the location of the VCF file used as a set of known
sites. You can include multiple collections of known sites by
repeating the
-k KNOWN_SITESoption. - RECALIBRATED_BAM: the location and filename of the recalibrated BAM output file. A corresponding index file (.bai) will be created. This output is optional as Sentieon® variant callers can perform the recalibration on the fly using the before recalibration BAM plus the recalibration table
2.2.5. Variant calling¶
A single command is run to call variants and additionally apply the BQSR calculated before.
sentieon driver -t NUMBER_THREADS -r REFERENCE -i DEDUPED_BAM \
-q RECAL_DATA.TABLE --algo Haplotyper [-d dbSNP] VARIANT_VCF
You may want to rerun only the variant calling, for example to use Unified Genotyper variant calling algorithm. In that case, you do not need to re-apply the BQSR, and can use the previously recalibrated BAM:
sentieon driver -t NUMBER_THREADS -r REFERENCE -i RECALIBRATED_BAM \
--algo Genotyper [-d dbSNP] VARIANT_VCF
In both cases, using the recalibrated BAM or the BAM before recalibration plus the recalibration data table will give identical results; however, you should be careful not to use the recalibration data table together with the already recalibrated BAM, as that would apply the recalibration twice, leading to incorrect results.
The following inputs are required for the command:
- NUMBER_THREADS: the number of computer threads that will be used in the calculation. We recommend that the number does not exceed the number of computing cores available in your system.
- REFERENCE: the location of the reference FASTA file. You should make sure that the reference is the same as the one used in the mapping stage.
- DEDUPED_BAM: the location where the previous deduping stage stored the result.
- RECAL_DATA.TABLE: the location where the previous BQSR stage stored the result.
- RECALIBRATED_BAM: the location of the recalibrated BAM file.
- VARIANT_VCF: the location and filename of the variant calling output file. A corresponding index file will be created. The tool will output a compressed file by using .gz extension.
The following inputs are optional for the command:
- dbSNP: the location of the Single Nucleotide Polymorphism database (dbSNP) that will be used to label known variants. You can only use one dbSNP file.