
4. Typical usage for TNseq®¶
Another of the typical uses of Sentieon® Genomics software is to perform the bioinformatics pipeline for Tumor-Normal analysis recommended in the Broad institute Somatic short variant discovery (SNVs + Indels). Fig. 4.1 illustrates such a typical bioinformatics pipeline.
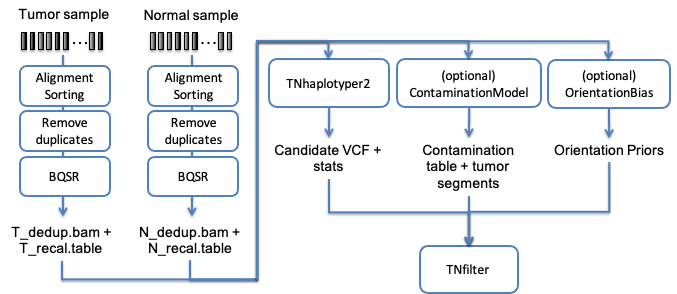
Fig. 4.1 Recommended bioinformatics pipeline for Tumor-Normal analysis
Please check the appnote in https://support.sentieon.com/appnotes/somatic for recommended somatic variant calling pipelines with different inputs.
4.1. General¶
In this bioinformatics pipeline you will need the following inputs:
- The FASTA file containing the nucleotide sequence of the reference genome corresponding to the sample you will analyze.
- Two sets of FASTQ files containing the nucleotide sequence of the sample to be analyzed, one for the tumor sample and one for the matched normal sample. These files contain the raw reads from the DNA sequencing. The software supports inputting FASTQ files compressed using GZIP. The software only supports files containing quality scores in Sanger format (Phred+33).
You can also include in the pipeline the following optional inputs that will help the algorithms detect artifacts and remove false positives:
- Panel of normal VCF: list of common errors that appear as variants from multiple unrelated normal samples. The contents of this file will be used to identify variants that are more likely to be germline variants, and filter them as such.
- Population resource VCF: list of population allele specific frequencies that will be used for filtering possible germline variants and to annotate the results.
The following steps compose the typical bioinformatics pipeline for a tumor-normal matched pair:
- Independently pre-process the Tumor and Normal samples using a DNAseq
pipeline like the one introduced in Section 2, with the following stages:
- Map reads to reference; you need to make sure that the SM sample tag is different between the tumor and the normal samples, as you will need it as an argument in the somatic variant calling.
- Calculate data metrics.
- Remove duplicates.
- (Optional) Base quality score recalibration (BQSR).
- Somatic variant discovery, with the following stages:
- (Optional) Estimate the cross-sample contamination and tumor segmentation.
- (Optional) Estimate any possible orientation bias present in the sequencing.
- Somatic candidate variant calling on the two individual BAM files: this step identifies the potential sites where the cancer genome data displays somatic variations relative to the normal genome, and calculates genotypes at that site.
- Filter the variants.
4.2. Step by step usage¶
For the mapping, duplicate removal, and base quality score recalibration stages, please refer to Section 2.2 for detailed usage instructions.
4.2.1. Variant discovery with matched normal sample¶
Two commands are run to call variants on the tumor-normal matched pair. We recommend that the optional steps of estimation of cross-sample contamination and estimation of orientation bias be run in the same command as TNhaplotyper to improve performance.
sentieon driver -t NUMBER_THREADS -r REFERENCE \
-i TUMOR_DEDUPED_BAM [-q TUMOR_RECAL_DATA.TABLE] \
-i NORMAL_DEDUPED_BAM [-q NORMAL_RECAL_DATA.TABLE] \
--algo TNhaplotyper2 --tumor_sample TUMOR_SAMPLE_NAME \
--normal_sample NORMAL_SAMPLE_NAME \
[--germline_vcf GERMLINE_RESOURCE] \
[--pon PANEL_OF_NORMAL] \
TMP_OUT_TN_VCF \
[ --algo OrientationBias --tumor_sample TUMOR_SAMPLE_NAME \
ORIENTATION_DATA ] \
[ --algo ContaminationModel --tumor_sample TUMOR_SAMPLE_NAME \
--normal_sample NORMAL_SAMPLE_NAME \
--vcf GERMLINE_RESOURCE \
--tumor_segments CONTAMINATION_DATA.segments \
CONTAMINATION_DATA ]
sentieon driver -r REFERENCE \
--algo TNfilter --tumor_sample TUMOR_SAMPLE_NAME \
--normal_sample NORMAL_SAMPLE_NAME \
-v TMP_OUT_TN_VCF \
[--contamination CONTAMINATION_DATA] \
[--tumor_segments CONTAMINATION_DATA.segments] \
[--orientation_priors ORIENTATION_DATA] \
OUT_TN_VCF
The following inputs are required for the command:
- NUMBER_THREADS: the number of computer threads that will be used in the calculation. We recommend that the number does not exceed the number of computing cores available in your system.
- REFERENCE: the location of the reference FASTA file. You should make sure that the reference is the same as the one used in the mapping stage.
- TUMOR_DEDUPED_BAM: the location of the pre-processed BAM file after deduplication for the TUMOR sample.
- NORMAL_DEDUPED_BAM: the location of the pre-processed BAM file after deduplication for the NORMAL sample.
- TUMOR_SAMPLE_NAME: sample name used for tumor sample in Map reads to reference stage.
- NORMAL_SAMPLE_NAME: sample name used for normal sample in Map reads to reference stage.
- TMP_OUT_TN_VCF: the location and file name of the output file from TNhaplotyper2; this is a temporary file.
- OUT_TN_VCF: the location and file name of the output file containing the variants.
The following inputs are optional for the command:
- TUMOR_RECAL_DATA.TABLE: the location where the BQSR stage for the TUMOR sample stored the result.
- NORMAL_RECAL_DATA.TABLE: the location where the BQSR stage for the NORMAL sample stored the result.
- PANEL_OF_NORMAL: the location and name of panel of normal VCF file.
- GERMLINE_RESOURCE: the location of the population germline resource. The AF INFO field in the resource file will be used to annotate variant alleles with the population allele frequencies, which will then be used to identify possible germline variants that are not true somatic variants.
- ORIENTATION_DATA: the location and file name of the file containing the orientation bias information produced by OrientationBias.
- CONTAMINATION_DATA: the location and file name of the file containing the contamination information produced by ContaminationModel.
- CONTAMINATION_DATA.segments: the location and file name of the file containing the tumor segments information produced by ContaminationModel.
4.2.2. Special considerations when missing a matched normal sample¶
When missing a matched normal sample, we recommend the following changes to the pipeline shown in the previous section:
- The PANEL_OF_NORMAL and GERMLINE_RESOURCE should be included, as they replace the missing matched normal sample. Missing both the matched normal and these germline resources will result in a large number of germline false positive calls in the final output VCF.
- The --normal_sample argument and the input BAM and recalibration table for the normal sample should not be included.
4.2.3. Generating GERMLINE_RESOURCE germline population resource¶
The germline population resource file containing the population allele frequencies can be obtained from gnomAD by post-processing the file following this procedure:
Download all per chromosome .vcf.bgz files for the version you want.
For each chromosome, remove unnecessary annotations to reduce file size:
bcftools annotate -x ^INFO/AF,INFO/AC INPUT.chrN.vcf.bgz | bcftools norm -m +any -Oz -o OUTPUT.chrN.vcf.gzConcatenate all files onto a single file, and create the corresponding index:
bcftools concat -Oz -o OUTPUT.vcf.gz OUTPUT.chr*.vcf.gz && bcftools index -t OUTPUT.vcf.gz
For instance, to download the GnomAD v3.1 release from Google, you can run:
for chr in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X Y; do curl https://storage.googleapis.com/gcp-public-data--gnomad/release/3.1/vcf/genomes/gnomad.genomes.v3.1.sites.chr$chr.vcf.bgz \ | bcftools annotate -x ^INFO/AF,INFO/AC - | bcftools norm -m +any -Oz -o tmp_OUTPUT.chr$chr.vcf.gz file_list="$file_list tmp_OUTPUT.chr$chr.vcf.gz" done bcftools concat -Oz -o OUTPUT.vcf.gz $file_list && bcftools index -t OUTPUT.vcf.gz rm $file_list