
Copy Number Variant¶
Introduction¶
This document describes the typical usage of Copy Number Variant (CNV) caller. If you have any additional questions, please contact the technical support at Sentieon® Inc. at support@sentieon.com.
Typical usage of the CNV¶
Sentieon® Genomics
software is able to perform the bioinformatics pipeline for Copy Number
Variant (CNV) discovery recommended by Broad Institute as in
http://gatkforums.broadinstitute.org/gatk/discussion/9143. cnv-fig1
below illustrates such a typical bioinformatics pipeline. cnv-fig2 shows
the pipeline for creating CNV Panel of Normals.
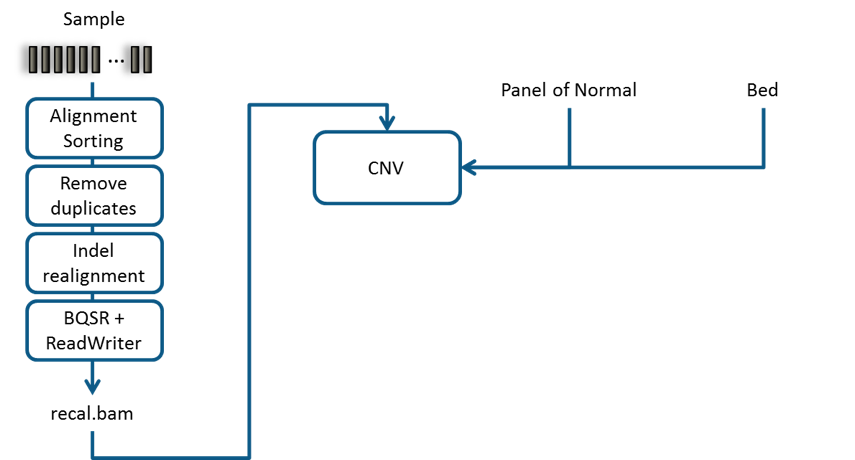
Recommended bioinformatics pipeline for CNV
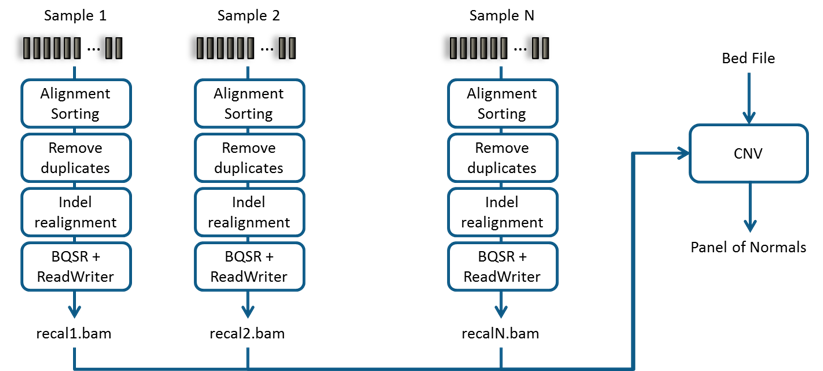
Recommended bioinformatics pipeline for creating CNV Panel of Normals
General¶
In this bioinformatics pipeline, you will need the following inputs:
- The FASTA file containing the nucleotide sequence of the reference specimen corresponding to the sample you will use.
- Independently pre-process the sample using a DNAseq pipeline with the
following stages:
- Map reads to reference.
- Calculate data metrics.
- Remove duplicates.
- Indel realignment (optional).
- Base quality score recalibration (BQSR).
- Panel of Normals (PoN) built using the PoN pipeline described below.
Panel of Normal (PoN) is created using the following input:
- The FASTA file containing the nucleotide sequence of the reference specimen corresponding to the sample you will use.
- Independently pre-process a large cohort of samples (recommend >40)
using a DNA seq pipeline with the following stages:
- Map reads to reference.
- Calculate data metrics.
- Remove duplicates.
- Indel realignment (optional).
- Base quality score recalibration (BQSR).
- The bed file defines the probes for coverage in the sequenced regions
for whole-exome data, or target regions spanning the whole genome for
whole-genome data. Please note:
- Beside the three required columns for Bed file (chromosome name, start and end positions in the chromosome), the target name is also required in the fourth column. Target names must be unique in the bed file.
- The probe window size defines the granularity and sensitivity of the CNV event detection.
- The target information saved in the PoN will be used for future CNV calling
Typical usage¶
CNV variant discovery¶
A single command is run to call CNV on a single-sample BAM input:
sentieon driver -t NUMBER_THREADS -r REFERENCE -i RECALED_BAM --algo CNV --pon PON_FILE OUT_CNV
The following inputs are required for the command:
- NUMBER_THREADS: the number of computer threads that will be used in the calculation. We recommend that the number does not exceed the number of computing cores available in your system.
- REFERENCE: the location of the reference FASTA file. You should make sure that the reference is the same as the one used in the mapping stage.
- RECALED_BAM: the location and filename of the re-calibrated BAM file from BQSR stage, or uncalibrated BAM with BQSR table. As noted before, we only support input BAM file with only one sample.
- PON_FILE: location of the file containing Panel of Normals collected from a large cohort of similarly prepared samples.
- OUT_CNV: the location and file name of the output file containing the variants.
Create Panel of Normals (PoN)¶
A single command is run to create Panel of normal:
sentieon driver -t NUMBER_THREADS -r REFERENCE -i RECALED_BAM [-i RECALED_BAM] \
--algo CNV --target BED_FILE [--target_padding PADDING] [--target_min_split MIN_PROBE_SIZE] \
--create_pon OUT_PON
The following inputs are required for the command:
- NUMBER_THREADS: the number of computer threads that will be used in the calculation. We recommend that the number does not exceed the number of computing cores available in your system.
- REFERENCE: the location of the reference FASTA file. You should make sure that the reference is the same as the one used in the mapping stage.
- RECALED_BAM: the location and filename of the re-calibrated BAM files from BQSR stage, or uncalibrated BAM with BQSR table. Based on Broad Institute’s recommendation, at least 40 samples are needed to create a reliable PoN.
- BED_FILE: the location and filename of the BED file containing the interested regions.
- OUT_PON: the location and file name of the output PoN file. The PoN file is in HDF5 format.
The following inputs are optional for the command:
- PADDING: Number of base-pairs extended from each end of the intervals in the bed file. Default: 250.
- MIN_PROBE_SIZE: Min probe size in number of base-pairs as long targets are split. Default: 500. When this value is set to -1, target merging and splitting is disabled.
The input targets will undergo the following steps depending on the value of target_min_split:
- When target_min_split is not -1, all targets will be padded, merged, and re-split into new targets with sizes between MIN_PROBE_SIZE and 2*MIN_PROBE_SIZE.
- When target_min_split = -1, the user is responsible for resolving target overlaps, and split long targets to reasonable and roughly uniform lengths. In this case, targets will the padded based on user input, and the overlaps resulting from padding will be resolved.
A file at the location of OUT_PON with the filename OUT_PON.targetWeights is also created. This file can be used as the input for updating the created PoN, as described in the next.
Update Panel of Normals (PoN)¶
A single command is run to update Panel of normal:
sentieon driver -t NUMBER_THREADS -r REFERENCE -i RECALED_BAM [-i RECALED_BAM] \
--algo CNV --coverage COVERAGE_FILE --create_pon OUT_PON
Beside the required and optional inputs as in Create Panel of Normals (PoN), when updating the Panel of Normals, the user also needs to input the following:
- COVERAGE: the location and file name of the coverage file. This file is created when creating the original PoN, at the same location as the PoN, but with a suffix .targetWeights appended to PoN filename.
Please note that target input, including target file, padding and minimum split size, is no longer needed. The target information contained in the coverage file will be used instead.
The final PoN will contain the samples from the original coverage file, plus those in the input BAM for this command.
Appendix¶
This section describes how to create, update the intermediate target coverage file with the CNV tool, and how to use it to either create Panel of Normals (PoN), or call copy number variant.
Create/update target coverage file¶
A single command is run to create target coverage file:
sentieon driver -t NUMBER_THREADS -r REFERENCE -i RECALED_BAM [-i RECALED_BAM] \
--algo CNV --target BED_FILE [--target_padding PADDING --coverage COVERAGE_FILE] \
--create_coverage OUT_COVERAGE
The meaning of the input argument is the same as above, except that:
--create_coverageargument allows user to only generate the target coverage file.- With
--coverage COVERAGE_FILE, the target coverage from the input samples will be appended to those in COVERAGE_FILE. The final result will be saved to OUT_COVERAGE.
Please note that when coverage file is specified, the redundant target input, including target file, padding and minimum split size, should not be specified.
Use target coverage file to create PoN¶
A single command is run to create Panel of Normals (PoN):
sentieon driver -t NUMBER_THREADS -r REFERENCE --algo CNV --coverage COVERAGE_FILE --create_pon OUT_PON
The meaning of the input argument is the same as above, except that:
- No BAM input files are specified.
--coverageargument is required to provide the target coverage data to create PoN.
Use target coverage file to call CNV¶
A single command is run to create CNV variant:
sentieon driver -t NUMBER_THREADS -r REFERENCE --algo CNV --pon PON_FILE --coverage COVERAGE_FILE OUT_CNV
The meaning of the input argument is the same as above, except that:
- No BAM input files are specified.
--coverageargument is required to provide the target coverage data for CNV variant discovery.- COVERAGE_FILE should only contain coverage for one sample.
Please note that identical target setting should be used to create the target coverage and PoN files.