
Unique Molecular Identifiers¶
Introduction¶
Attention
Support for UMI tags has been added to the LocusCollector and Dedup
algos in the Sentieon driver. Usage of the LocusCollector and Dedup
algos with UMI tags is described in the app note at,
https://support.sentieon.com/appnotes/PCRdedup/.
We recommend new pipelines use the LocusCollector and Dedup algos
for UMI-aware consensus read creation instead of the approach described in
this appnote.
This document describes using the Sentieon® tools to process next-generation sequence data while taking advantage of molecular barcode information (also called unique molecular indices or UMIs). Molecular barcodes introduce unique tags on the ends of template DNA molecules prior to sequencing to greatly reduce the impact of PCR duplicates and sequencing errors on the variant calling process.
The Sentieon® tools provide functionality for extracting UMI tags from read data and performing barcode-aware consensus generation. This pipeline expects adapter-free barcoded reads as input. The output of the UMI consensus pipeline is a BAM file containing consensus molecules derived from the barcoded read data. These consensus molecules can be used as input to most variant calling software.
Sentieon® UMI pipeline¶
Overall UMI pipeline structure¶
Sentieon® provides two utilities for UMI NGS data processing:
umi extract: UMI tag extraction from unaligned input reads with adapter already removedumi consensus: Barcode-aware duplicate removal and consensus calling on aligned input
Sentieon® suggests the following typical UMI processing pipeline (umi-fig1):
- UMI tag extraction from unaligned input reads with
umi extractutility- Alignment to the reference genome with Sentieon®
bwa mem- UMI consensus calling with
umi consensusutility- Alignment and sorting of consensus reads to the reference genome with Sentieon®
bwa mem.
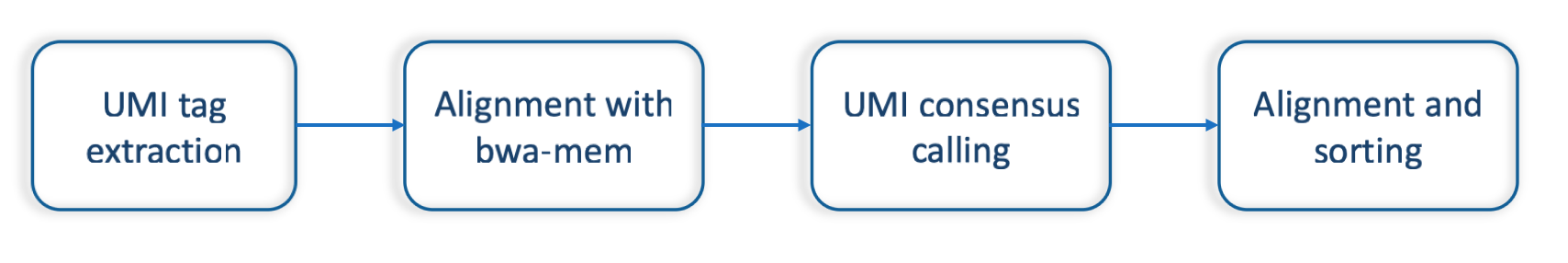
Sentieon® UMI processing pipeline
Below is a code example. Details of each component will be explained in the subsequent sections.
sentieon umi extract \
8M12S+T,+T \
sample_R1.fastq.gz \
sample_R2.fastq.gz | \
sentieon bwa mem \
-R "@RG\tID:$GROUP\tSM:$SAMPLE\tLB:$LIBRARY\tPL:$PLATFORM" \
-t $NT \
-K $BWA_K_SIZE \
-p \
-C \
$REF \
- | \
sentieon umi consensus \
-o sample_consensus.fastq.gz
sentieon bwa mem \
-R "@RG\tID:$GROUP\tSM:$SAMPLE\tLB:$LIBRARY\tPL:$PLATFORM" \
-t $NT \
-K $BWA_K_SIZE \
-p \
-C \
$REF \
sample_consensus.fastq.gz | \
sentieon util sort \
-i - \
-o sample_consensus.bam \
--sam2bam --umi_post_process
Determine the read structure and extract barcode sequences¶
As a first step, you need to extract the barcode sequences from the input reads.
This is performed with Sentieon® umi extract command, which
extracts the barcode sequence information from the reads and adds it to the read
description. As noted earlier, the adapter sequence should be removed from the input
reads before running umi tag extraction. This can be performed by other third-party tools.
The output of umi extract is in FASTQ format with interleaved R1 and R2 reads. By default,
the output from the extract command will be sent to standard output unless otherwise defined with the option -o.
The syntax of the umi extract command is as follows:
sentieon umi extract [options] read_structure fastq1 [fastq2] [fastq3]
Options:
-o Output file (default: stdout)
-d Turn on duplex mode
--umi_tag Logic umi tag (default 'XR')
--output_format Output format FASTQ or SAM (default 'FASTQ')
The first argument of umi extract command defines the read structure. For paired-end reads,
two read structures separated by comma ',' should be specified.
A read structure is defined by <number><operator> pairs. The number can be any digit or '+'
to indicate the end of the read. Possible operators include:
- T The template sequence.
- B The cell barcode sequence.
- M The molecular barcode sequence.
- S A sequence of bases that should be ignored.
The -d option is used to extract duplex UMI and label the strand it originates.
Duplex UMI extraction requires an identical read structure for both strands.
As an example, the following command demonstrates a single-ended UMI extraction
on pair-ended reads. In this case, the first read in the pair
contains an 8bp molecular barcode followed by a 12bp spacer and then
the template sequence. The second read contains only the template sequence. The
paired reads will be interleaved in the output file. Please note that in this example
the output is piped to gzip to generate compressed FASTQ file. In general, we
recommend piping the output directly to the next step (Sentieon® bwa mem).
sentieon umi extract "8M12S+T,+T" \
sample_R1.fastq.gz \
sample_R2.fastq.gz | \
gzip -c \
> sample_extracted_pair.fastq.gz
The command below demonstrates duplex UMI extraction where both reads contain 4bp molecular barcode followed by template sequence.
sentieon umi extract \
-d \
"4M+T,4M+T" \
sample_R1.fastq.gz \
sample_R2.fastq.gz | \
gzip -c \
> sample_extracted_pair.fastq.gz
Below is a use case when the UMI sequence is already in a separate FASTQ file sample_I1.fastq.gz.
When running in this mode, only one additional UMI index read is allowed. The UMI index read should
contain no template sequences. This mode does not allow duplex UMI extraction.
sentieon umi extract \
"+M,+T,+T" \
sample_I1.fastq.gz \
sample_R1.fastq.gz \
sample_R2.fastq.gz | \
gzip -c \
> sample_extracted_pair.fastq.gz
The command below produces a SAM format output. This format is useful when using the
RNA-seq aligner STAR. In this example, the first read in the pair contains a 16bp
cell barcode and a 10bp molecular barcode. The second read contains only the template
sequence. The output is single-end reads in the SAM format.
sentieon umi extract \
--output_format SAM \
"16B10M+S,+T" \
sample_R1.fastq.gz \
sample_R2.fastq.gz \
> sample_extracted.sam
The output of umi extract contains additional tags.
By default, the output contains CR tag for cell or sample barcode, and XR tag
for UMI sequence to be used by umi consensus.
| Tags | Meaning |
|---|---|
| RX | Extracted UMI sequence bases. |
| XR | UMI tag for grouping in umi consensus. |
| CR | Cell barcode. |
Alignment to the reference genome¶
The interleaved fastq can be aligned to the reference genome using bwa mem. The
-p option is necessary to specify that the input file is an interleaved
fastq while the -C option will cause the barcode tags in the fastq
description to be appended to the read's SAM record.
sentieon bwa mem \
-R "@RG\tID:$GROUP\tSM:$SAMPLE\tLB:$LIBRARY\tPL:$PLATFORM" \
-t $NT \
-K $BWA_K_SIZE \
-p \
-C \
$REF \
sample_extracted_pair.fastq.gz > sample_aligned.sam
Consensus molecule creation¶
The next stage of the pipeline is creating consensus molecules from aligned
barcode-tagged reads using Sentieon® umi consensus.
The syntax of umi consensus is as follows:
umi consensus [-i input] [options] -o output
Options for umi_consensus:
-i, --input Input file (default: stdin SAM)
-o, --output Output file
--input_format SAM/BAM/CRAM
--umi_tag Logic UMI tag (default:'XR')
--copy_tags List of tags to be copied (default: XR,RX,MI,BI,BD,XZ)
--read_name_prefix
Read name prefix (default: 'UMI-')
By default, umi consensus will read input from the standard input with SAM format.
This can be overriden with --input option that defines the input file, and
--input_format option for the file format other than SAM.
Output from umi consensus is a an interleaved fastq containing consensus molecules,
which can be re-mapped by Sentieon® bwa mem. Below is example:
cat sample_aligned.sam | \
sentieon umi consensus \
-o sample_consensus.fastq.gz
The output of umi consensus generates the following additional tags.
| Tags | Meaning |
|---|---|
| BI/BD | Indel quality scores |
| MI | A unique label for UMI group where the consensus is derived. |
| XZ | The number of raw reads within the UMI group where the consensus is derived. For duplex UMI this will contain the number of raw reads from each strand. |
Report consensus reads without the BI/BD tags¶
By default, umi consensus recalibrates the INDEL error rates and store such information
in the BI/BD tags. This modeling step can be turned off by removing the BI/BD tags
from the --copy_tags option.
cat sample_aligned.sam | \
sentieon umi consensus \
--copy_tags XR,RX,MI,XZ \
-o sample_consensus.fastq.gz
Alignment of consensus reads to the reference genome¶
The interleaved fastq from umi consensus can be aligned to the reference genome
using Sentieon® bwa mem. Similar to the previous alignment, the -p option and the
-C option are necessary. Piping the output to Sentieon® util sort creates the output
BAM file ready for variant calling. The --umi_post_process option is used to instruct
the tool to perform the necessary post-processing of the consensus reads.
sentieon bwa mem \
-R "@RG\tID:$GROUP\tSM:$SAMPLE\tLB:$LIBRARY\tPL:$PLATFORM" \
-t $NT \
-K $BWA_K_SIZE \
-p \
-C \
$REF \
sample_consensus.fastq.gz | \
sentieon util sort \
-i - \
-o sample_consensus.bam \
--sam2bam --umi_post_process
Variant calling from consensus reads¶
The BAM file from UMI consensus pipeline is analysis ready for variant calling. Additional steps of duplicate marking or base quality adjustments should not be performed because the UMI consensus step is essentially a combination of both PCR duplicate removal and base quality recalibration.
Although any somatic caller can be used with the consensus reads, we recommend TNscope® because of its high sensitivity for low-frequency variant detection.
Logs from UMI consensus¶
The log printed by umi consensus contains statistical information on the input reads that
can be useful for quality control. The two pieces of information currently available are:
Group size histogram: Group size is the number of supporting raw reads within a group that call one consensus read. The histogram shows the number of UMI consensus reads that have a specific group size and can be used to detect issues:
- If the proportion of singletons (UMI consensus with group size = 1) is too high, it will be difficult to accurately model the PCR error rates and improve the quality scores.
- Conversely, if the average reads per UMI group is too high, it could be an indication that the amount of input DNA in the library preparation was too low, which in turn will lead to low coverage of consensus reads.
Duplex stat: With duplex UMI tags,
umi consensusis able to recognize reads originating from each strand of DNA by comparing the UMI tags from both R1 and R2 reads, and identify errors raised during sample preparation. The numbers reported in the log represent the number of consensus reads that are either single strand or duplex, grouped by the following codes:
Group code 1: Single-strand consensus with no reads from the complementary UMI group.
Group code 2: Duplex consensus with reads from two complementary UMI groups.
Group code 3: UMI sequences from R1 and R2 reads are the same. In this case,
umi consensusrelies on the strand of inserts to determine whether the input reads are from the same strand. This group is further divided into:
- Group code 31: Single-strand consensus with all reads coming from the same strand.
- Group code 32: Duplex consensus with reads from both strands of DNA.