
Distributed Mode¶
Introduction¶
This document describes how to use the sharding capabilities of the Sentieon® Genomics tools to distribute a DNAseq® pipeline onto multiple servers; distribution of other pipelines such as a TNseq® one follow the same principles, as all Sentieon® genomcis tools have the same built in distribution processing capabilities. The goal of such distribution is to reduce the overall runtime of the pipeline to produce results quicker; however such distribution has some overhead that makes the computation cost higher.
Using the distribution capabilities, each stage of the pipeline is divided into small tasks; each of these tasks processes part of the genome, and can be run in different servers in parallel. Each of these tasks produces a partial result that needs to be merged sequentially into a final single output; such merging needs to be carefully done to make sure it considers the boundary and produces the same results as a pipeline run without sharding.
The execution framework for the distribution is not in the scope of this document, and the user needs to distribute the data/files and launch the correct processes while keeping the correct data dependency among the stages.
Shard and sharding¶
We divide the genome into many sequential non-overlapping parts, with each part called a shard. Each shard is defined as either a single chromosome contig name, or part of a chromosome following the convention contig:shard_start-shard_end. The special shard NO_COOR is used for all unmapped reads that do not have coordinate.
The Sentieon® binaries support sharding for distribution into multiple
servers, and can process multiple shards in a single command by adding
one or multiple shard options with the --shard SHARD argument. When
using multiple --shard options in a single command line, these shards
need to be contiguous according to the reference contig list; for
example, a command can have one shard covering the end of chr2 and one
shard covering the beginning of chr3, but it should not have a shard
covering the end of chr2 and the beginning of chr22.
You can refer to the generate_shards.sh script in the appendix for a sample
file that creates shards for the genome based on either the dictionary
associated with the reference or the bam header in the input bam files.
We recommend using 100 M bases as the shard size.
Data flow and data/file dependency for distribution¶
A pipeline for DNAseq® following the recommended workflow processes a pair of fastq files through the following stages: BWA alignment to produce a sorted.bam, Deduplication to produce a dedup.bam, BQSR to produce a recal.table and Haplotyper to produce an output.vcf.gz file. Fig. 1 illustrates the data flow for such a pipeline.
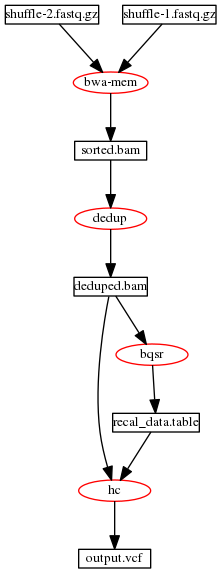
Fig. 1 Typical data flow for DNAseq® pipeline
To distribute the above pipeline onto multiple servers, each of the stages is divided into commands that process the data on a shard, requiring inputs from both the specific shard as well as the right and left neighboring shards; the exceptions to this are the Dedup stage, which requires ALL the score files from the LocusCollector commands on all shards, and the Haplotyper stages, which requires a complete merged recalibration table.
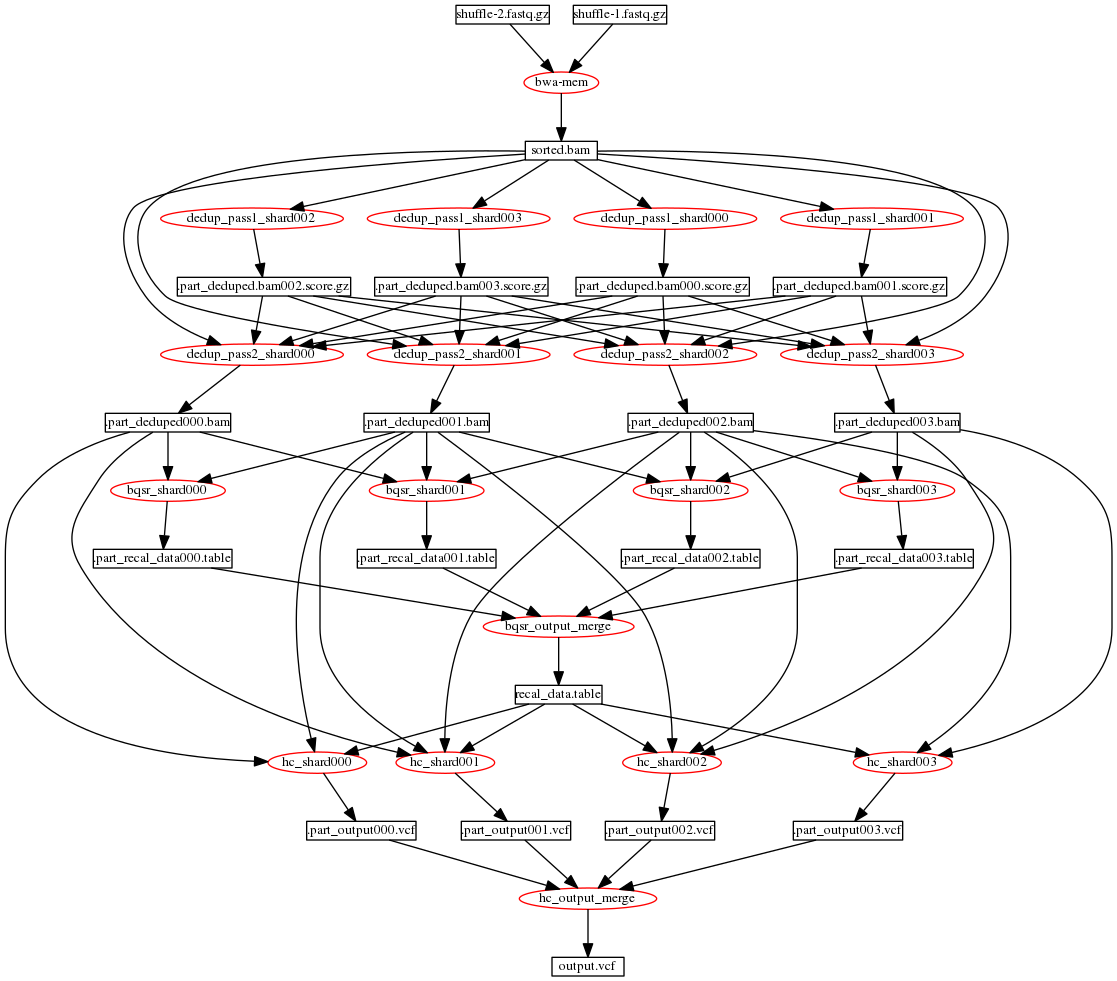
Fig. 2 Data flow for DNAseq® pipeline distributed as 4 shards without processing unmapped reads
As an example, Fig. 2 illustrates the data flow for a pipeline distributed as 4 shards; this is not a typical use case as using the recommended shard size of 100M bases will result in requiring more than 30 shards. In the example of Fig. 2, the stages require the following inputs and produce the following outputs:
- The LocusCollector stage (dedup pass 1) for the
i-thshard requires the sorted.bam input. The stage produces apart_deduped.bam$shard_i.score.gzfile. - The Dedup stage (dedup pass 2) for the
i-thshard requires the sorted.bam input as well as all the results from all the LocusCollector stages. The stage produces apart_deduped$shard_i.bamfile. - The QualCal stage for the
i-thshard requires thepart_deduped$shard_i.bamfile as well as thepart_deduped$shard_i+1.bamfile andpart_deduped$shard_i-1.bamfile if available. The stage produces apart_recal_data$shard_i.tablefile. - After QualCal, all
part_recal_data$shard_i.tablefiles need to be merged into a single calibration table file, to be used in variant calling. The options--passthruin the driver and--mergeof QualCal can be used to perform the boundary aware merging. - The Haplotyper stage for the
i-thshard requires thepart_deduped$shard_i.bamfile as well as thepart_deduped$shard_i+1.bamfile andpart_deduped$shard_i-1.bamfile if available; additionally, the complete merged recalibration table needs to be an input. The stage produces apart_output$shard_i.vcf.gzfile - After Haplotyper, all
part_output$shard_i.vcf.gzfiles need to be merged into a single output VCF file. The options--passthruin the driver and--mergeof Haplotyper are used to perform the boundary aware merging. - If at any stage in the process you require to have the merged output
bam file, you can use the util binary to perform the boundary aware
merging; you need to add the option
--mergemode=10in the command so that util merge does not process the reads and only copies them by block.
Important
When merging the results, be careful to input the partial results sequentially in the correct order, as otherwise the results will be incorrect.
Distributing BWA¶
The above instructions do not include any information about distributing
the alignment using BWA. In order to distribute the BWA alignment, you
can use the fqidx tool contained in the Sentieon® tools to create index
files for the input FASTQ files; then you can extract certain parts of
the FASTQ files for processing on different servers, using the results
of the fqidx command as the input to BWA mem; you will need to make sure
that the option -p is included in the BWA command, as the output of the
fqidx contains interleaved reads in a single output. The results of this
flow are identical to executing on a single run.
BWA_K_size=100000000
num_splits=10
SAMPLE="sample_name" #Sample name SM tag in bam
GROUP="read_group_name" #Read Group name RGID tag in bam
PLATFORM="ILLUMINA"
NT=$(nproc) #number of threads to use in computation, set to all threads available
FASTA="/home/regression/references/b37/human_g1k_v37_decoy.fasta"
FASTQ_1="WGS-30x_1.fastq.gz"
FASTQ_2="WGS-30x_2.fastq.gz"
FASTQ_INDEX="WGS-30x.fastq.gz.index"
#create FASTQ indices
sentieon fqidx index -K $BWA_K_size -o $FASTQ_INDEX $FASTQ_1 $FASTQ_2
#get the number of runs that the inputs will be split into given the size
num_K=$(sentieon fqidx query -i $FASTQ_INDEX | cut -d' ' -f1)
BWA_K_size=$(sentieon fqidx query -i $FASTQ_INDEX | cut -d' ' -f2)
num_K_splits=$(expr $num_K / $num_splits + 1)
#run multiple BWA on multiple servers
file_list=""
for run in $(seq 0 $((num_splits-1))); do
region="$((run*num_K_splits))-$((run*num_K_splits+num_K_splits))"
#send each of these to a different server
sentieon bwa mem -R "@RG\tID:$GROUP\tSM:$SAMPLE\tPL:$PLATFORM" \
-K $BWA_K_size -t $NT -p $FASTA \
"<sentieon fqidx extract -i $FASTQ_INDEX -r $region $FASTQ_1 $FASTQ_2" | \
sentieon util sort -r $FASTA -t $NT --sam2bam -o sorted_run$run.bam -i -
file_list="$file_list sorted_run$run.bam"
done
#merge the results
sentieon util merge -o sorted.bam $file_list
#or perform deduplication on all the intermediate BAM files
file_list_bam=""
for file in $file_list; do file_list_bam="$file_list_bam -i $file"; done
sentieon driver -r $FASTA -t $NT $file_list --algo LocusCollector --fun score_info score.txt.gz
sentieon driver -r $FASTA -t $NT $file_list --algo Dedup --score_info score.txt.gz deduped.bam
Distributing BWA when you cannot create FASTQ index file (version 201808.02+)¶
In the event that it is not feasible to create the FASTQ index files,
you can use util fqidx with the fractional option -F m/n while also using the
-K option. This divides the input FASTQs in splits of n chuncks of reads and
then extracts every m-th element from each split to be processed in a different
server, with m going from 0 to n-1.
Bear in mind that this feature is only recommended in an IT environment where the file storage is fast enough to support each fqidx process reading the input FASTQ files at the same time, which is common in cloud environments, or in local cluster environments with a high bandwidth NFS system.
BWA_K_size=100000000
num_splits=10
SAMPLE="sample_name" #Sample name SM tag in bam
GROUP="read_group_name" #Read Group name RGID tag in bam
PLATFORM="ILLUMINA"
NT=$(nproc) #number of threads to use in computation, set to all threads available
FASTA="/home/regression/references/b37/human_g1k_v37_decoy.fasta"
FASTQ_1="WGS-30x_1.fastq.gz"
FASTQ_2="WGS-30x_2.fastq.gz"
#run multiple BWA on multiple servers
file_list=""
for run in $(seq 0 $((num_splits-1))); do
#send each of these to a different server
sentieon bwa mem -R "@RG\tID:$GROUP\tSM:$SAMPLE\tPL:$PLATFORM" \
-K $BWA_K_size -t $NT -p $FASTA \
"<sentieon fqidx extract -F $((run))/$num_splits -K $BWA_K_size $FASTQ_1 $FASTQ_2" | \
sentieon util sort -r $FASTA -t $NT --sam2bam -o sorted_run$run.bam -i -
file_list="$file_list sorted_run$run.bam"
done
#merge the results
sentieon util merge -o sorted.bam $file_list
#or perform deduplication on all the intermediate BAM files
file_list_bam=""
for file in $file_list; do file_list_bam="$file_list_bam -i $file"; done
sentieon driver -r $FASTA -t $NT $file_list --algo LocusCollector --fun score_info score.txt.gz
sentieon driver -r $FASTA -t $NT $file_list --algo Dedup --score_info score.txt.gz deduped.bam
Distributing GVCFtyper for large cohort joint calling¶
For large joint calls with more than 1000 samples, we recommend setting
--genotype_model multinomial in GVCFtyper. While the default genotyping mode
is theoretically more accuracy for smaller cohorts, the multinomial mode is
equally accurate with large cohorts and scales better with a very large numbers
of samples.
When running large number of samples (>3000) joint cohort calling, distribution using similar techniques as the ones described above are recommended; however, a few challenges need to be taken into account:
- The overall runtime of the joint calling.
- The resource requirements of the machines where the distributed GVCFtyper command will be run.
- The logistics of storing and accessing the large number of GVCF input files.
- The file size of the output VCF file.
In general, the recommendation is to use the Sentieon® sharding
capabilities of GVCFtyper to process different genomic shards in
different machines. The commands below access the complete list of GVCF
inputs, and assume those inputs are available in a common location such
as a NFS storage or a remote object storage location accessible either
through s3 or http/https protocols:
#individual shard calling in machine 1
sentieon driver -r $FASTA -t $NT --shard $shard1 --shard $shard2 \
--algo GVCFtyper $gvcf_argument GVCFtyper-shard_1.vcf.gz
#individual shard calling in machine 2
sentieon driver -r $FASTA -t $NT --shard $shard3 \
--algo GVCFtyper $gvcf_argument GVCFtyper-shard_2.vcf.gz
…
Important
The list of input GVCFs needs to be ordered consistently when processing each shard, as the sample order in the final output will depend on the input order and merging will require all partial files to have the same sample order.
The output VCF files from GVCFtyper when using --shard option are not valid VCF files, so they should not be used until you merge them as described below.
After all shards are processed, you need to run a GVCFtyper merge
command to merge the results, making sure that the order of the
intermediate VCF inputs is consistent with the reference. The input files
need to be available in a common location such as a NFS storage or a
remote object storage location accessible either through s3 or http/https
protocols:
#merge step
sentieon driver --passthru -t $nt --algo GVCFtyper --merge joint_final.vcf.gz \
GVCFtyper-shard_1.vcf.gz GVCFtyper-shard_2.vcf.gz …
Overall runtime and resource requirements¶
In order to reduce the overall runtime, it is recommended to use enough shards to allow granular distribution of the runtime into multiple servers. The shards can be created either by shard size or expected complexity. However, the final merge step of the individual shard results cannot be distributed and needs to be run in a single server; this fact sets a lower bound on the number of servers to be used for the distribution, as the merging could dominate the overall runtime.
The GVCFtyper algorithm is a very efficient algorithm that will likely be I/O limited by the performance of the storage location of the GVCF inputs. As such, it is recommended to store the GVCF inputs in a file system that provides 600 MBps transfer rate.
For extremely large number of samples (>10000) joint cohort calling, memory may become a problem and certain OS restrictions may also come into play. For such a case, it is recommended to do the following:
Set the OS open file limit to a big enough number, to allow the software to open enough file handles. This is done via the command
ulimit -n NUM.Set the OS stack limit to a big enough number, to allow the software to allocate enough memory for the operation, as the memory is proportional to the number of samples, and may be too big to fit in the stack allocated by default. This is done via the command
ulimit -s NUM.Use a file containing the list of input GVCFs, to prevent the command to be longer that the argument list OS length. You can do this by using the following command:
sentieon driver ... --algo GVCFtyper output.vcf.gz - < input_files.txt
Use jemalloc memory allocation as described in the jemalloc appnote at, https://support.sentieon.com/appnotes/jemalloc/ and update the vcf cache size by adding the following code to your scripts:
export VCFCACHE_BLOCKSIZE=4096
Set the shard size to a smaller number, i.e. 50MBases. In addition, use the driver option
--traverse_paramin the GVCFtyper commands to make sure that all threads are fully utilized. The command would become:sentieon driver -r $FASTA -t $NT --shard $shard1 --shard $shard2 \ --traverse_param 10000/200 \ --algo GVCFtyper GVCFtyper-shard_1.vcf.gz - < input_files.txt
Challenge of the large output VCF file¶
When running a very large cohort, the output VCF file will contain a very large number of columns with the genotype information of each sample. This large number of columns may make the output file unwieldy, for instance making running VQSR on the full file impractical, so you may need to look into alternatives to VCF storage of the output.
Depending on the method you will use to store the output, splitting the output by either sample groups or genomic coordinates may ameliorate the issue of very large output files; please feel free to contact sentieon support with your specific request on how you will store the output of joint calling.
Splitting output by genomic regions¶
In order to make the output VCF file smaller, you can perform the shard merging across specific genomic sub-regions, for instance across individual chromosomes. You would do this by merging only a subset of the intermediate VCF files. For example you can create per chromosome VCF files by merging only the shards that cover each chromosome: if shards 1-4 cover chr1 and shard 5 covers both chr1 and chr2, the following code would create a VCF containing only chr1 vartiants:
#merge the necessary intermediate sharded VCFs
sentieon driver --passthru -t $nt --algo GVCFtyper --merge \
GVCFtyper_chr1_tmp.vcf.gz \
GVCFtyper-shard_1.vcf.gz GVCFtyper-shard_2.vcf.gz GVCFtyper-shard_3.vcf.gz \
GVCFtyper-shard_4.vcf.gz GVCFtyper-shard_5.vcf.gz
#remove variants not in the proper contig and index the VCF
bcftools view GVCFtyper_chr1_tmp.vcf.gz -r chr1 \
-o - | sentieon util vcfconvert - GVCFtyper_chr1.vcf.gz
Using the above methodology allows you to create valid VCF files with a fraction of the size of the full VCF.
In order to run VQSR on the output above, you need to supply the VarCal algorithm
with a VCF file containing all variant records across the complete genome, as they
are required to perform the proper calibration of the VQSR model. However, VarCal
only requires the first 8 columns of VCF data, so you do not need to concatenate all
the VCF from each specific genomic sub-regions and can create a smaller VCF file that
contains the necesary information by extracting and concatenating the first 8 columns
of each file. Using the code below will create the necessary file:
vcf_list=(GVCFtyper_chr1.vcf.gz GVCFtyper_chr2.vcf.gz) # include more VCFs if applicable
#extract the first 8 columns from each region to a new compressed VCF
mkfifo tmp.fifo
sentieon util vcfconvert tmp.fifo GVCFtyper_annotations.vcf.gz &
convert_pid=$!
exec 3>tmp.fifo #a file descriptor to hold the fifo open
bcftools view -h ${vcf_list[0]} | grep "^##" > tmp.fifo
bcftools view -h ${vcf_list[0]} | grep "^#CHROM" | cut -f 1-8 > tmp.fifo
for vcf in ${vcf_list[@]}; do
bcftools view -H $vcf | cut -f 1-8 > tmp.fifo
done
exec 3>&-
wait $convert_pid
rm tmp.fifo
The VarCal algo can then be run on the merged VCF containing the first eight
columns of the VCF to generate a tranches and recal file for both SNPs and
INDELs. VQSR can then be applied directly to the VCFs split by genomic region
with the ApplyVarCal algo:
vcf_list=(GVCFtyper_chr1.vcf.gz GVCFtyper_chr2.vcf.gz) # include more VCFs if applicable
#apply variant quality score recalibration
for vcf in ${vcf_list[@]}; do
out_vcf=${vcf%%.vcf.gz}_snp-indel-recal.vcf.gz
sentieon driver -r $FASTA -t $nt --algo ApplyVarCal -v $vcf \
--vqsr_model var_type=SNP,tranches_file=${snp_tranches},sensitivity=99.0,recal=${snp_recal} \
--vqsr_model var_type=INDEL,tranches_file=${indel_tranches},sensitivity=99.0,recal=${indel_recal} \
$out_vcf
done
Splitting output by sample groups¶
Alternativelly, the Sentieon® GVCFtyper merge includes the --split_by_sample
algo option as a potential solution to deal with this challenge. The
--split_by_sample algo option is used during the merge step to generate valid
partial VCFs, each containing a subset of samples, thus separating the complete
VCF file into smaller, more manageable output files. The usage of the
--split_by_sample algo option is:
sentieon driver --passthru -t $nt --algo GVCFtyper --merge \
--split_by_sample split.conf GVCFtyper_main.vcf.gz \
GVCFtyper-shard_1.vcf.gz GVCFtyper-shard_2.vcf.gz …
The split.conf input file used in the --split_by_sample algo option is
a tab separated file where each line starts with the output file of the
specific sample group, followed by a tab separated list of the samples
of the corresponding group. You can use more than one line with the same
output file, which will group all samples from the multiple lines. The
example below shows two groups of samples where samples 1 to 5 will be
output to the group1 output file, while samples 6 to 8 will be output to
the group2 output file:
GVCFtyper_file_group1.vcf.gz Sample1 Sample2 Sample3
GVCFtyper_file_group1.vcf.gz Sample4 Sample5
GVCFtyper_file_group2.vcf.gz Sample6 Sample7 Sample8
The GVCFtyper merge command above will generate the following outputs:
- GVCFtyper_main.vcf.gz: a partial VCF file containing all the VCF information up to and including the INFO column. No sample information will be included. This file is useful to run VQSR, as it contains all the necessary information for the variant recalibration.
- GVCFtyper_file_group1.vcf.gz: a partial VCF file containing the up to and including the INFO column, plus the FORMAT column, and all the columns for the samples including in group1.
- GVCFtyper_file_group2.vcf.gz: a partial VCF file containing the up to and including the INFO column, plus the FORMAT column, and all the columns for the samples including in group2.
You can then use bcftools to merge the partial VCFs and select the samples you are interested in. You can use the following code to do it:
bash extract.sh GVCFtyper_main.vcf.gz \
split.conf Samle1,Sample4,Sample7
Where the 3rd argument of the script is a comma separated list of the samples of interest and the extract.sh script is:
#!/bin/bash
MAIN=$1
GRPS_CONF=$2
SAMPLES=$3
REGIONS=$4
BCF=/home/release/other_tools/bcftools-1.3/bcftools
if [ $# -eq 4 ] || [ $# -eq 3 ]; then
if [ $REGIONS == "" ]; then
BED_ARG=""
else
BED_ARG="-R $REGIONS"
fi
#parse input files from group config file
GRPS=$(grep -v "^#" $GRPS_CONF| cut -f1 | sort | uniq)
$BCF view -h $MAIN | grep -v '^#CHROM' | grep -v '^##bcftools' > out.vcf
hdr=$($BCF view -h $MAIN | grep '^#CHROM')
hdr="$hdr\tFORMAT"
arg="<($BCF view $BED_ARG -H -I $MAIN | cut -f -8)"
col=9
for g in $GRPS; do
s=$($BCF view --force-samples -s $SAMPLES -h $g 2>/dev/null | grep '^#CHROM' | \
cut -f 10-)
[ -z "$s" ] && continue
hdr="$hdr\t$s"
c="<($BCF view --force-samples -s $SAMPLES $BED_ARG -H -I $g 2>/dev/null | \
cut -f $col-)"
arg="$arg $c"
col=10
done
echo -e "$hdr" >> out.vcf
eval paste "$arg" >> out.vcf
else
echo "usage $0 main_vcf_file group_config_file csv_sample_list [bed_file]"
fi
The solution presented here allows to easily run VQSR on the GVCFtyper_main.vcf.gz to create a file after recalibration, which you can then use in the extract.sh script to obtain the after VQSR results.
Special considerations for Cloud environments¶
In cloud environments, there is typically no NFS storage that can accommodate a large number of GVCF inputs. For joint genotyping, the Sentieon® GVCFtyper allows GVCF input files hosted in an object storage location such as AWS s3 or in a location accessible via HTTP. However, for large cohorts (100+), the memory requirements for the GVCFtyper command using object storage inputs will likely be too high, so this method of accessing the inputs is not recommended.
The recommended methodology in a cloud environment is to download partial GVCF input files to the computing node depending on the shard that the node is processing. The partial download of the GVCF inputs can be done using bcftools, but it is important to add the option --no-version to the bcftools command to make sure that the headers of the different shards are not different enough to trigger the GVCFtyper merge from rejecting them:
#use bcftools to download shard1 GVCF partial inputs and create the index
bcftools view --no-version -r $shard1_csv_intervals -o - $URL_sample.g.vcf.gz | \
sentieon util vcfconvert - ${sample}_s1.g.vcf.gz
#or download multiple files in parallel using xargs using a list
cat list_inputs.txt | xargs -P $NUM_PARALLEL_DOWNLOAD -I @ | \
sh -c "bcftools view -r $shard1_csv_intervals -o - $S3BUCKET_INPUTS/@ | \
sentieon util vcfconvert - @"
To gain additional efficiency, a “waterfall” approach can be used, as shown in Fig. 3 and Fig. 4. In this approach a computing node will process multiple shards sequentially, running the download of the partial GVCF inputs for the next shard in parallel with the GVCFtyper processing of the current shard, thus more efficiently sharing resources as a CPU intensive and I/O intensive process would. The pipeline would be as follows:
- Download the partial GVCF inputs for shard1.
- Start the GVCFtyper of shard1, and in parallel download the partial GVCF inputs for shard2.
- After the previous step is completed, start the GVCFtyper of shard2, and in parallel use bcftools to download shard3.
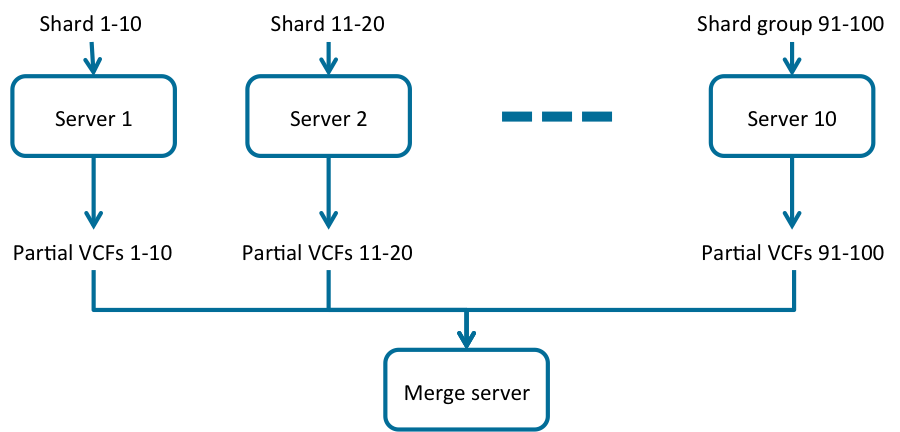
Fig. 3 Distributing GVCFtyper onto multiple servers in a cloud environment
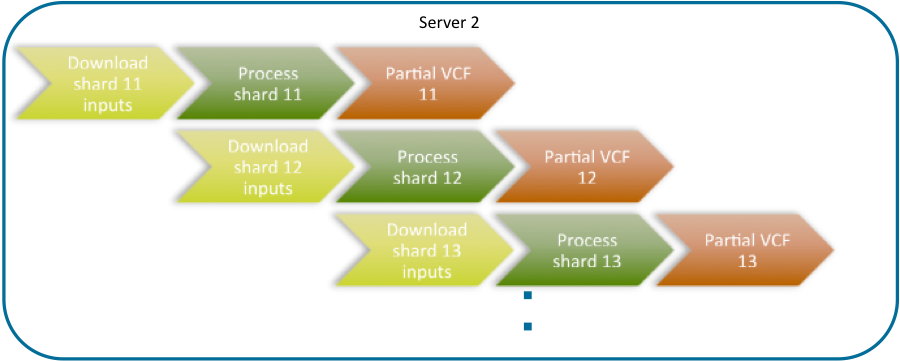
Fig. 4 Distributing GVCFtyper onto multiple servers in a cloud environment, detail of the waterfall approach of downloading the inputs of the next shard in parallel with the processing of the previous shard
Shell examples¶
Below is a shell example for the distribution commands, using shard size 1G bases, which was selected for demo purposes only. The recommended shard size is 100M bases.
# Sample file for distributing DNAseq pipeline onto 4 1GBase shards
# Each stage command can be distributed to a different server for faster processing,
# but the user needs to make sure that the necessary files are present in each machine
FASTA="/home/b37/human_g1k_v37_decoy.fasta"
FASTQ_1="WGS-30x_1.fastq.gz"
FASTQ_2="WGS-30x_2.fastq.gz"
FASTQ_INDEX="WGS-30x.fastq.gz.index"
KNOWN1="/home/b37/1000G_phase1.indels.b37.vcf.gz"
KNOWN2="/home/b37/Mills_and_1000G_gold_standard.indels.b37.vcf.gz"
DBSNP="/home/b37/dbsnp_138.b37.vcf.gz"
#######################################
# BWA mapping, distributed on 4 servers
#######################################
BWA_K_size=100000000
num_srvr=4
#get the number of runs that the inputs will be split into given the size
num_K=$(sentieon fqidx query -i $FASTQ_INDEX | cut -d' ' -f1)
BWA_K_size=$(sentieon fqidx query -i $FASTQ_INDEX | cut -d' ' -f2)
num_K_srvr=$(expr $num_K / $num_srvr + 1)
#run multiple BWA on multiple servers
file_list=""
for run in $(seq 0 $((num_srvr-1))); do
region="$((run*num_K_srvr))-$((run*num_K_srvr+num_K_srvr))"
#send each of these to a different server
sentieon bwa mem -R "@RG\tID:$GROUP\tSM:$SAMPLE\tPL:$PLATFORM" \
-K $BWA_K_size -t $NT -p $FASTA \
"< sentieon fqidx extract -i $FASTQ_INDEX -r $region $FASTQ_1 $FASTQ_2" | \
sentieon util sort -r $FASTA -t $NT --sam2bam -o sorted_run$run.bam -i -
file_list="$file_list sorted_run$run.bam"
done
#merge the results
sentieon util merge -o sorted.bam $file_list
#######################################
# define 4 shards
#######################################
SHARD_0="--shard 1:1-249250621 --shard 2:1-243199373 --shard 3:1-198022430 \
--shard 4:1-191154276 --shard 5:1-118373300"
SHARD_1="--shard 5:118373301-180915260 --shard 6:1-171115067 --shard 7:1-159138663 \
--shard 8:1-146364022 --shard 9:1-141213431 --shard 10:1-135534747 \
--shard 11:1-135006516 --shard 12:1-49085594"
SHARD_2="--shard 12:49085595-133851895 --shard 13:1-115169878 --shard 14:1-107349540 \
--shard 15:1-102531392 --shard 16:1-90354753 --shard 17:1-81195210 \
--shard 18:1-78077248 --shard 19:1-59128983 --shard 20:1-63025520 \
--shard 21:1-48129895 --shard 22:1-51304566 --shard X:1-118966714"
SHARD_3="--shard X:118966715-155270560 --shard Y:1-59373566 --shard MT:1-16569 \
--shard GL000207.1:1-4262 --shard GL000226.1:1-15008 --shard GL000229.1:1-19913 \
--shard GL000231.1:1-27386 --shard GL000210.1:1-27682 --shard GL000239.1:1-33824 \
--shard GL000235.1:1-34474 --shard GL000201.1:1-36148 --shard GL000247.1:1-36422 \
--shard GL000245.1:1-36651 --shard GL000197.1:1-37175 --shard GL000203.1:1-37498 \
--shard GL000246.1:1-38154 --shard GL000249.1:1-38502 --shard GL000196.1:1-38914 \
--shard GL000248.1:1-39786 --shard GL000244.1:1-39929 --shard GL000238.1:1-39939 \
--shard GL000202.1:1-40103 --shard GL000234.1:1-40531 --shard GL000232.1:1-40652 \
--shard GL000206.1:1-41001 --shard GL000240.1:1-41933 --shard GL000236.1:1-41934 \
--shard GL000241.1:1-42152 --shard GL000243.1:1-43341 --shard GL000242.1:1-43523 \
--shard GL000230.1:1-43691 --shard GL000237.1:1-45867 --shard GL000233.1:1-45941 \
--shard GL000204.1:1-81310 --shard GL000198.1:1-90085 --shard GL000208.1:1-92689 \
--shard GL000191.1:1-106433 --shard GL000227.1:1-128374 \
--shard GL000228.1:1-129120 --shard GL000214.1:1-137718 \
--shard GL000221.1:1-155397 --shard GL000209.1:1-159169 \
--shard GL000218.1:1-161147 --shard GL000220.1:1-161802 \
--shard GL000213.1:1-164239 --shard GL000211.1:1-166566 \
--shard GL000199.1:1-169874 --shard GL000217.1:1-172149 \
--shard GL000216.1:1-172294 --shard GL000215.1:1-172545 \
--shard GL000205.1:1-174588 --shard GL000219.1:1-179198 \
--shard GL000224.1:1-179693 --shard GL000223.1:1-180455 \
--shard GL000195.1:1-182896 --shard GL000212.1:1-186858 \
--shard GL000222.1:1-186861 --shard GL000200.1:1-187035 \
--shard GL000193.1:1-189789 --shard GL000194.1:1-191469 \
--shard GL000225.1:1-211173 --shard GL000192.1:1-547496 \
--shard NC_007605:1-171823 --shard hs37d5:1-35477943"
#######################################
# Locus collector
#######################################
#Locus Collector for shard 000
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -i sorted.bam $SHARD_0 \
--algo LocusCollector --fun score_info .part_deduped.bam000.score.gz \
2> collect000.log
#Locus Collector for shard 001
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -i sorted.bam $SHARD_1 \
--algo LocusCollector --fun score_info .part_deduped.bam001.score.gz \
2> collect001.log
#Locus Collector for shard 002
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -i sorted.bam $SHARD_2 \
--algo LocusCollector --fun score_info .part_deduped.bam002.score.gz \
2> collect002.log
#Locus Collector for shard 003
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -i sorted.bam $SHARD_3 \
--algo LocusCollector --fun score_info .part_deduped.bam003.score.gz \
2> collect003.log
#Locus Collector for shard with unmapped reads (NO_COOR)
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -i sorted.bam --shard NO_COOR \
--algo LocusCollector --fun score_info .part_deduped.bam004.score.gz \
2> collect004.log
#######################################
# Dedup using all score.gz files
#######################################
#Dedup for shard 000
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -i sorted.bam $SHARD_0 \
--algo Dedup --score_info .part_deduped.bam000.score.gz \
--score_info .part_deduped.bam001.score.gz \
--score_info .part_deduped.bam002.score.gz \
--score_info .part_deduped.bam003.score.gz \
--score_info .part_deduped.bam004.score.gz \
--rmdup .part_deduped000.bam 2> dedup000.log
#Dedup for shard 001
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -i sorted.bam $SHARD_1 \
--algo Dedup --score_info .part_deduped.bam000.score.gz \
--score_info .part_deduped.bam001.score.gz \
--score_info .part_deduped.bam002.score.gz \
--score_info .part_deduped.bam003.score.gz \
--score_info .part_deduped.bam004.score.gz \
--rmdup .part_deduped001.bam 2> dedup001.log
#Dedup for shard 002
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -i sorted.bam $SHARD_2 \
--algo Dedup --score_info .part_deduped.bam000.score.gz \
--score_info .part_deduped.bam001.score.gz \
--score_info .part_deduped.bam002.score.gz \
--score_info .part_deduped.bam003.score.gz \
--score_info .part_deduped.bam004.score.gz \
--rmdup .part_deduped002.bam 2> dedup002.log
#Dedup for shard 003
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -i sorted.bam $SHARD_3 \
--algo Dedup --score_info .part_deduped.bam000.score.gz \
--score_info .part_deduped.bam001.score.gz \
--score_info .part_deduped.bam002.score.gz \
--score_info .part_deduped.bam003.score.gz \
--score_info .part_deduped.bam004.score.gz \
--rmdup .part_deduped003.bam 2> dedup003.log
#Dedup for shard with unmapped reads (NO_COOR)
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -i sorted.bam --shard NO_COOR \
--algo Dedup --score_info .part_deduped.bam000.score.gz \
--score_info .part_deduped.bam001.score.gz \
--score_info .part_deduped.bam002.score.gz \
--score_info .part_deduped.bam003.score.gz \
--score_info .part_deduped.bam004.score.gz \
--rmdup .part_deduped004.bam 2> dedup004.log
#Merge bam files from all shards into final output
$SENTIEON_FOLDER/bin/sentieon util merge -i .part_deduped000.bam \
-i .part_deduped001.bam -i .part_deduped002.bam \
-i .part_deduped003.bam -i .part_deduped004.bam \
-o deduped.bam --mergemode=10 2> dedup_merge.log
#######################################
# BQSR
#######################################
#QualCal for shard 000
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -r $FASTA -i .part_deduped000.bam \
-i .part_deduped001.bam $SHARD_0 --algo QualCal -k $KNOWN1 -k $KNOWN2 \
.part_recal_data000.table 2> bqsr000.log
#QualCal for shard 001
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -r $FASTA -i .part_deduped000.bam \
-i .part_deduped001.bam -i .part_deduped002.bam $SHARD_1 --algo QualCal \
-k $KNOWN1 -k $KNOWN2 .part_recal_data001.table 2> bqsr001.log
#QualCal for shard 002
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -r $FASTA -i .part_deduped001.bam \
-i .part_deduped002.bam -i .part_deduped003.bam $SHARD_2 --algo QualCal \
-k $KNOWN1 -k $KNOWN2 .part_recal_data002.table 2> bqsr002.log
#QualCal for shard 003
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -r $FASTA -i .part_deduped002.bam \
-i .part_deduped003.bam $SHARD_3 --algo QualCal -k $KNOWN1 -k $KNOWN2 \
.part_recal_data003.table 2> bqsr003.log
#QualCal for shard with unmapped reads (NO_COOR)
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -r $FASTA -i .part_deduped004.bam \
--shard NO_COOR --algo QualCal -k $KNOWN1 -k $KNOWN2 \
.part_recal_data004.table 2> bqsr004.log
#Merge table files into complete calibration table
$SENTIEON_FOLDER/bin/sentieon driver --passthru --algo QualCal \
--merge recal_data.table .part_recal_data000.table .part_recal_data001.table \
.part_recal_data002.table .part_recal_data003.table .part_recal_data004.table \
2> bqsr_merge.log
#######################################
# Variant Calling
#######################################
#Haplotyper for shard 000
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -r $FASTA -i .part_deduped000.bam \
-i .part_deduped001.bam -q recal_data.table $SHARD_0 --algo Haplotyper \
-d $DBSNP .part_output000.vcf.gz 2> hc000.log
#Haplotyper for shard 001
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -r $FASTA -i .part_deduped000.bam \
-i .part_deduped001.bam -i .part_deduped002.bam -q recal_data.table \
$SHARD_1 --algo Haplotyper -d $DBSNP .part_output001.vcf.gz 2> hc001.log
#Haplotyper for shard 002
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -r $FASTA -i .part_deduped001.bam \
-i .part_deduped002.bam -i .part_deduped003.bam -q recal_data.table \
$SHARD_2 --algo Haplotyper -d $DBSNP .part_output002.vcf.gz 2> hc002.log
#Haplotyper for shard 003
$SENTIEON_FOLDER/bin/sentieon driver -t 16 -r $FASTA -i .part_deduped002.bam \
-i .part_deduped003.bam -q recal_data.table $SHARD_3 --algo Haplotyper \
-d $DBSNP .part_output003.vcf.gz 2> hc003.log
#There is no need to do variant calling on the NO_COOR shard, as there will
# be no variants on unmapped reads
#Merge output files into output VCF
$SENTIEON_FOLDER/bin/sentieon driver --passthru --algo Haplotyper \
--merge output.vcf.gz .part_output000.vcf.gz .part_output001.vcf.gz \
.part_output002.vcf.gz .part_output003.vcf.gz 2> hc_merge.log
Below is a shell example generate_shards.sh to create the shards.
determine_shards_from_bam(){
local bam step tag chr len pos end
bam="$1"
step="$2"
pos=1
samtools view -H $bam |\
while read tag chr len; do
[ $tag == '@SQ' ] || continue
chr=$(expr "$chr" : 'SN:\(.*\)')
len=$(expr "$len" : 'LN:\(.*\)')
while [ $pos -le $len ]; do
end=$(($pos + $step - 1))
if [ $pos -lt 0 ]; then
start=1
else
start=$pos
fi
if [ $end -gt $len ]; then
echo -n "$chr:$start-$len "
pos=$(($pos-$len))
break
else
echo "$chr:$start-$end"
pos=$(($end + 1))
fi
done
done
echo "NO_COOR"
}
determine_shards_from_dict(){
local bam step tag chr len pos end
dict="$1"
step="$2"
pos=1
cat $dict |\
while read tag chr len UR; do
[ $tag == '@SQ' ] || continue
chr=$(expr "$chr" : 'SN:\(.*\)')
len=$(expr "$len" : 'LN:\(.*\)')
while [ $pos -le $len ]; do
end=$(($pos + $step - 1))
if [ $pos -lt 0 ]; then
start=1
else
start=$pos
fi
if [ $end -gt $len ];then
echo -n "$chr:$start-$len "
pos=$(($pos-$len))
break
else
echo "$chr:$start-$end"
pos=$(($end + 1))
fi
done
done
echo "NO_COOR"
}
determine_shards_from_fai(){
local bam step tag chr len pos end
fai="$1"
step="$2"
pos=1
cat $fai |\
while read chr len other; do
while [ $pos -le $len ]; do
end=$(($pos + $step - 1))
if [ $pos -lt 0 ]; then
start=1
else
start=$pos
fi
if [ $end -gt $len ]; then
echo -n "$chr:$start-$len "
pos=$(($pos-$len))
break
else
echo "$chr:$start-$end"
pos=$(($end + 1))
fi
done
done
echo "NO_COOR"
}
if [ $# -eq 2 ]; then
filename=$(basename "$1")
extension="${filename##*.}"
if [ "$extension" = "fai" ]; then
determine_shards_from_fai $1 $2
elif [ "$extension" = "bam" ]; then
determine_shards_from_bam $1 $2
elif [ "$extension" = "dict" ]; then
determine_shards_from_dict $1 $2
fi
else
echo "usage $0 file shard_size"
fi